

Education
Technology


Recent Posts
Education
Technology
How SDSC and CENIC Are Bringing AI Infrastructure to California's Classrooms
By centralizing expert operations while allowing colleges to own their hardware, SDSC and CENIC AIR are providing equitable access to advanced...
Science
Technology
Using NSF ACCESS Supercomputers to Improve Tuberculosis Treatment Options
Researchers simulated an unprecedented number of tuberculosis drug combinations that may cure infections faster, using less medicine and reducing...
Technology
Science
Education
Using AI to Protect America's Farms from Frost
A California Central Valley initiative held a competition inviting university teams to develop data-driven models capable of predicting frost...

Technology
Science
Cheaper, Longer-Lasting Batteries Are Closer Thanks to a Pinch of Sodium and a Supercomputer
SDSC's Expanse played an important role in helping researchers design next gen sodium batteries that could make large‑scale energy...

Science
Technology
SDSC’s Expanse Helps Illustrate How to Break ‘Forever Chemicals’ in Real Time
Researchers used Expanse to simulate the atomic-level destruction of PFAS chemicals on electrically charged surfaces, thus providing a roadmap to...

Education
Science
Technology
Seeking Leaders for SDSC CORE Institute AI & Indigenous Language Revitalization Cohort
The leadership council will help shape an interdisciplinary cohort focused on building responsible, community-centered approaches to AI in...

Science
Technology
Education
SDSC Interns Drive Innovation in Alzheimer’s Research Data Tools
Student developers recently created a custom database web application that brings new efficiency and collaboration to a program once reliant on...

Science
Technology
SDSC’s Expanse Charts the Hidden Toll of Sickle Cell Disease
Patients face elevated risks of stroke, organ damage and chronic pain driven in part by this ongoing vascular injury. Using Expanse, researchers...

Education
Science
Technology
UC San Diego Students Learn HPC Skills via SDSC’s Expanse
At UC San Diego, high performance computing is being woven directly into undergraduate engineering education by giving students early, hands-on...
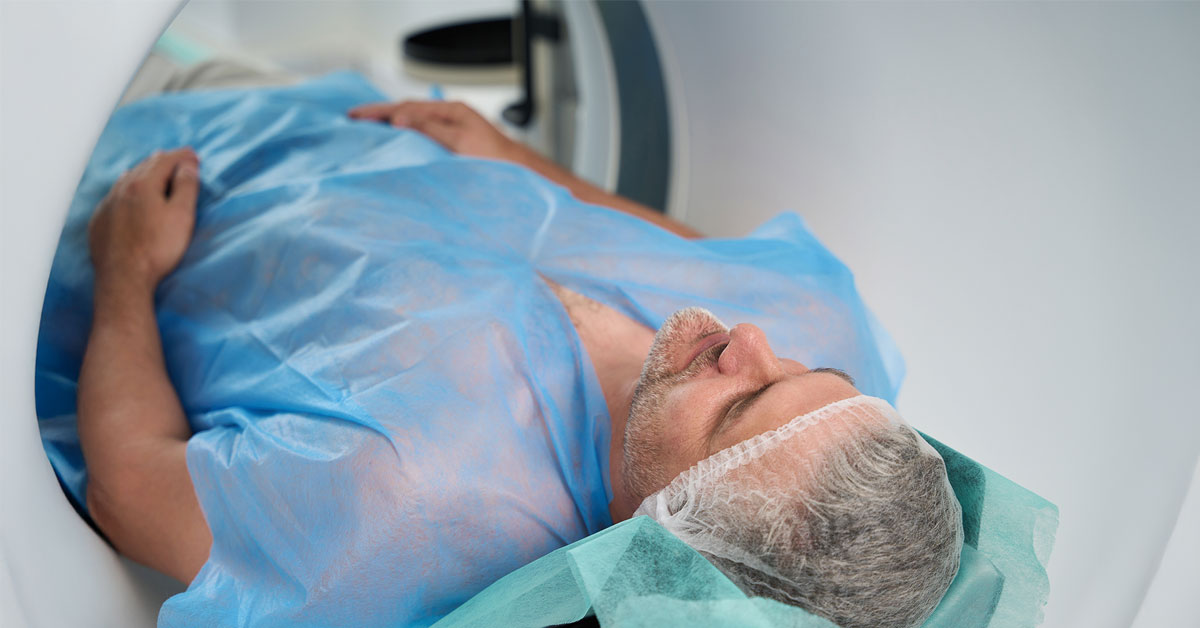
Science
Technology
San Diego Supercomputer Center Powers AI Model to Improve Prostate Cancer Care
A new AI model of the male urinary tract could make prostate cancer radiation therapy more precise and help reduce side effects, such as urinary...

Science
Technology
MIT Scientists Use SDSC Supercomputer to Reveal Hidden Physics Inside Metals
Researchers uncovered a hidden atomic process that challenges long-held assumptions about disorder in metals when they are melted, cooled or shaped.

Education
Science
Technology
SDSC Opens Summer Research Program for Local High School Students
SDSC’s REHS program gives participants hands‑on experience in data‑driven research while working alongside researchers and...

Technology
Science
Empowering Researchers to Decode Complex Biological Systems
From decoding microbial ecosystems to analyzing vast genetic datasets, modern biology increasingly relies on high-powered software tools that can...

Technology
Science
Hydrogen Power Gets a Spark — Thanks to SDSC’s Expanse
Solar panels and wind turbines increasingly dot the landscape, but the future of clean energy may well depend on how smoothly we burn hydrogen.

Education
Technology
Science
Farm-to-Table via Supercomputer
Across California, researchers, farmers, technologists and universities are launching a bold vision for the future of farming: “precision...

Technology
Science
AI Tool Promises Faster, More Accurate Cervical Cancer Treatment Planning
Researchers have developed a groundbreaking “one-click” AI tool that could transform how doctors plan specialized radiation treatments...

Technology
SDSC’s CloudBank: Empowering Recovery in Weather-Ravaged Communities
With support from CloudBank — a service that provides access to commercial cloud resources — the CIMA platform will deliver fast,...

Technology
Education
Congressional App Challenge Partners with SDSC to Strengthen the Nation’s Tech Workforce Pipeline
Several of San Diego County’s most promising high school coders recently visited SDSC to learn more about coding as part of the 2025...

Technology
SDSC Elevates and Ensures Secure Research Infrastructure for DOW Projects
The successful completion of the CMMC framework assessment marks SDSC as a trusted, fully compliant hosting platform for CUI and FCI, opening new...

Education
Technology
Awards
UC San Diego Team Takes Third Place at SC25 Student Cluster Competition in St. Louis
UC San Diego Team Sea++ was awarded third place at this year’s Student Cluster Competition, which took place at the International Conference...

Technology
Science
Cracking the Code of Parkinson’s: How Supercomputers Are Pointing to New Treatments
More than one million Americans live with tremors, slowed movement and speech changes caused by Parkinson’s disease — a degenerative...

Awards
SDSC Receives Honors in 2025 HPCwire Readers’ and Editors’ Choice Awards
The 22nd Annual HPCwire Awards were presented to SDSC leaders in the global HPC community.
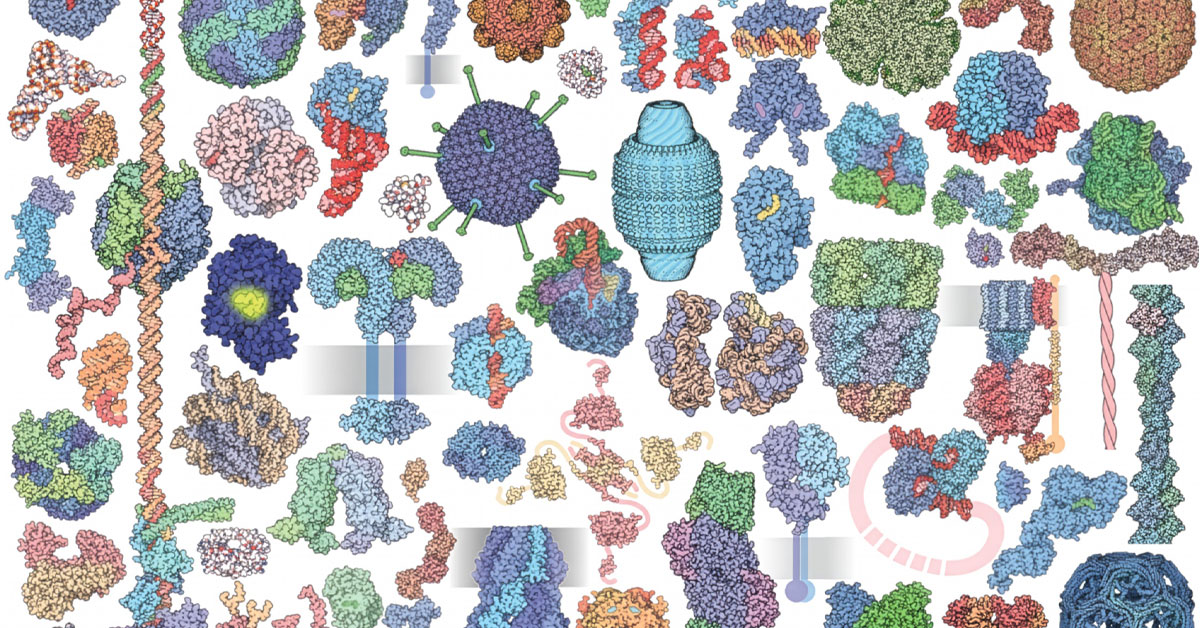
Technology
Science
SDSC-housed Protein Data Bank Brings Molecules Up to Size
Molecules craft the very fabric of reality while remaining too small for the human eye to glimpse. But thanks to PDB we can now “see”...

Technology
Education
Science
UC San Diego Team Joins Baltic Partners to Strengthen Ukraine's Groundwater Resilience and Workforce Development
The integration of remote sensing with machine learning allows researchers and students to create better predictions of groundwater storage and flows.

Technology
Education
CAIDA and Internet2 Collaborate for Internet Security Project to Benefit U.S. Research and Education Networks
Scientists share vast amounts of data across institutions and borders, but hidden internet routing problems can quietly send that data off course...